|
by Iain Beehuat Tan and Patrick Tan
 ancer of the stomach (gastric cancer) is the 4th most common cancer in the world and the 2nd leading cause of global cancer mortality, accounting for 8% of all newly-diagnosed cancer cases and 10% of cancer deaths worldwide. Particularly prevalent in several Asian countries including Japan, South Korea, China, and Singapore, most gastric cancer patients are diagnosed at advanced disease stages when surgery is no longer possible. In advanced cancer, several chemotherapy drugs have been demonstrated in clinical trials to provide tangible survival benefits, albeit modest. Clinicians have observed that patients with gastric cancer have different degree of disease aggressiveness and even when treated alike with the same treatment, have varying degrees of response with dramatic response and benefit with certain treatments and complete futility with other treatments. A central challenge in gastric cancer research has thus been to intelligently and rationally subdivide gastric cancers into homogenous subgroups that have similar clinical behavior, biological aberrations and molecular vulnerabilities and to devise optimized treatment strategies for each subgroup. ancer of the stomach (gastric cancer) is the 4th most common cancer in the world and the 2nd leading cause of global cancer mortality, accounting for 8% of all newly-diagnosed cancer cases and 10% of cancer deaths worldwide. Particularly prevalent in several Asian countries including Japan, South Korea, China, and Singapore, most gastric cancer patients are diagnosed at advanced disease stages when surgery is no longer possible. In advanced cancer, several chemotherapy drugs have been demonstrated in clinical trials to provide tangible survival benefits, albeit modest. Clinicians have observed that patients with gastric cancer have different degree of disease aggressiveness and even when treated alike with the same treatment, have varying degrees of response with dramatic response and benefit with certain treatments and complete futility with other treatments. A central challenge in gastric cancer research has thus been to intelligently and rationally subdivide gastric cancers into homogenous subgroups that have similar clinical behavior, biological aberrations and molecular vulnerabilities and to devise optimized treatment strategies for each subgroup.
Initial attempts in the mid-late 1990s to classify gastric cancers relied primarily on histology, where fixed sections of tumors are examined using light microscopy to identify differences in cellular morphology and nuclear architecture. In 1965, Lauren proposed a seminal histopathologic classification system for gastric cancer dividing gastric tumors into 2 main subtypes: intestinal-type and diffuse-type cancers. These 2 types looked distinct under the microscope. While intestinal-type tumors are largely composed of moderately to well differentiated gland-like structures, diffuse-type cancers are typified by solitary cells or small groups of tumour cells infiltrating normal gastric tissues, without formation of glands. Subsequent research (described below) has revealed that the 2 subtypes are likely to differ in the gender and age distribution of patients affected (epidemiology) and is associated with different sequence of events leading up from pre-cancerous disease to frank cancer (pathogenesis). More recent genetic analysis has also hinted that these subtypes may be characterized by distinct genetics both in terms of hereditary factors that are inherited (so called germline susceptibility patterns) or that are acquired in the process leading to formation of cancer (somatic genetic alterations). However, despite these potential differences, a significant degree of discordance also exists between different pathologists in classifying many gastric cancers as either intestinal and diffuse this has largely occurred because gastric cancers can sometimes exhibit mixed histologies bearing features of both subtypes. At present, the Lauren classification system is not considered by clinicians in deciding the management of individual gastric cancer patients. Existing classification systems have not enabled individual selection of chemotherapy. Beyond a small subset of gastric cancer patients (HER2 positive, described below), most gastric cancer patients today are still being treated with a one-size-fits-all strategy with markedly diverse clinical outcomes.
Alongside attempts by cancer researchers to classify gastric cancers, other researchers have also engaged in similar intensive efforts aimed at identifying genetic and environmental (non-genetic) risk factors influencing an individuals risk for developing gastric cancer. Currently, the single most established environmental risk factor for gastric cancer is infection by an infectious bacteria, Helicobacter pylori. Although the most common chronic bacterial infection in humans (50% of the world population is infected), fewer than 1% of people infected with H.pylori will actually develop gastric cancer. For these latter individuals, cancer risk is thought to be modulated by strain-specific virulence factors such as vacA, babA2, OipA and CagA, host responses and specific hostmicrobe interactions. Notably, while H.pylori is strongly associated with both intestinal-type and diffuse-type gastric cancers, the underlying carcinogenic mechanisms may be different. Specifically, in intestinal-type cancers H.pylori is implicated in a multi-step process beginning with early sequential changes called atrophic gastritis, intestinal metaplasia and dysplasia which ultimately lead to intestinal-type cancers. In contrast, diffuse-type cancers have no recognizable precursor lesions besides chronic gastritis. Importantly, eradication of H.pylori by antibiotics has clinically shown to attenuate gastric cancer risk, but only if atrophic gastritis and intestinal metaplasia have not yet occurred.
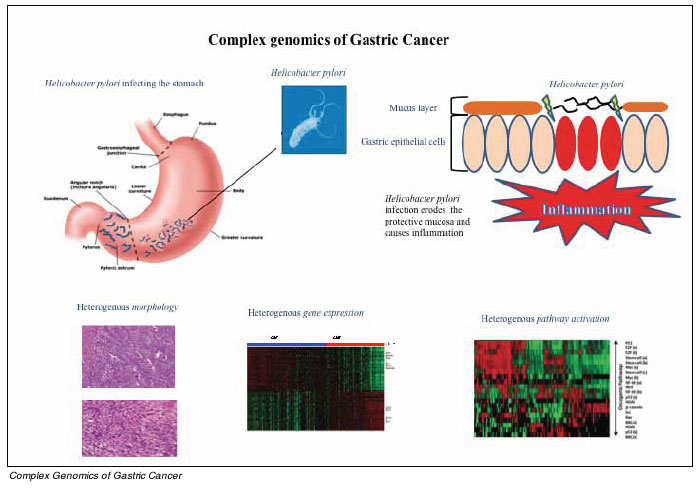
Since fewer than 1% of people infected with H.pylori will actually develop gastric cancer, additional factors that are present in the human host may influence gastric cancer risk (host factors). Indeed, genetic variation in genes (polymorphisms) involved in host inflammatory responses to infection such as cytokines, cytokine receptors and inflammation regulators have been associated with an increased risk of gastric cancer or precursor lesion formation. Genetic variability among regulators of stomach acidity (IL-1) and endothelial cell proliferation (IL-8) have also been implicated. One of the clearest examples, confirmed in several studies and meta-analyses, has been an association between increased susceptibility to H. pylori induced gastric cancer and specific single nucleotide polymorphisms in both interleukin 1ß and its endogenous receptor antagonist (IL-1RN). Besides genes related to inflammation, polymorphisms in mucin genes which are involved in establishing the gastric mucous layer protecting the stomach lining against proteases, mechanical damage, and pathogenic microorganisms like H. pylori, may also contribute to gastric cancer risk. Specifically, a gene dosage relationship has been observed with MUC1 and MUC5AC polymorphisms and an elevated risk of stomach cancer.
The advent of molecular genetics has also deepened our understanding of the molecular events leading to gastric cancer development. At the molecular level, 3 main mechanisms appear to be driving development of gastric cancer: chromosomal instability, microsatellite instability, and epigenetic alterations. These 3 processes are key to the accumulation of genomic damage in gastric epithelial cells, allowing them to acquire traits that comprise the hallmarks of cancer such as self-sufficiency in growth signals, resistance to apoptosis, replicative senescence, evasion of antigrowth signals, angiogenesis, and metastatic potential. While chromosomal instability, microsatellite instability and epigenetic alterations have been described in both Laurens subtypes, the relative contribution of these 3 processes and the specific genes affected are distinct. For example, p53 mutations are commonly found in intestinal-type gastric cancer and have also been observed even in intestinal metaplasia, suggesting that abrogation of normal p53 function appears to be important in intestinal-type cancers, Cell cycle genes are also frequently altered in intestinal-type cancers, with epigenetic silencing of p16 and p27 and/or amplification of Cyclin E1 being obseved. Mutations, loss of heterozygosity and methylation of APC are also frequently described in intestinal-type GCs, arguing for a role in its oncogenesis, perhaps in activating the WNT/ß-catenin signaling pathway. In contrast to intestinal-type cancers, sporadic diffuse-type GCs are associated with somatic CDH1 mutations, which interestingly tend to be splice site mutations.
Genes involved in the MAPK signaling pathway are also more frequently altered in intestinal-type cancers. Unlike other gastrointestinal cancers like colon or pancreatic cancers, K-RAS is infrequently mutated in gastric cancer, however those rare tumors with KRAS activating mutations are predominantly associated with the intestinal subtype. Perhaps one of the most important molecular contrasts between intestinal-type and diffuse-type gastric cancers lies in the gene amplification or overexpression of the HER2 growth factor receptor, as this difference directly influences patient management. In the landmark Phase 3 study (ToGA), the addition of Trastuzumab a monoclonal antibody targeting HER2, to palliative chemotherapy, led to an overall survival benefit specifically in patients with HER2 overexpression or amplification. As a result of this trial, Trastuzumab is now clinically approved as a treatment option for HER2 positive gastric cancers in several countries. Notably, around 20% of Intestinal-type cancers have HER2 amplification or overexpression compared to about 5% of Diffuse-type cancers. The success of ToGA underscores the critical need to identify new molecular subtypes of GC with underlying molecular vulnerabilities, which will enable the rational deployment of existing targeted agents or alternatively identify new druggable targets. For example, FGFR2, another growth factor receptor, has also been reported as overexpressed and amplified is also reported in gastric cancer. Recent pre-clinical work by our group and others suggests that FGFR2 targeting therapy be selectively effective in the setting of FGFR2-amplified gastric cancer.
Intestinal-type and diffuse-type gastric cancers also exhibit substantial abnormalities in their patterns of DNA aneuploidy (chromosome abnormalities), which can also impact on disease state and patient prognosis. Intestinal-type gastric cancers show a higher number of copy number gains at 8q, 17q, and 20q and losses at 3p and 5q. Diffuse-type gastric cancers show frequent copy number gains at 12q and 13q and losses at 4q, 15q, 16q, and 17p. In additional to broad chromosomal gains and losses, chromosomal instability may contribute to focal gene amplifications and losses. The role of HER2 amplification, particularly frequent in intestinal-type gastric cancers, has been discussed. Chromosomal instability also drives carcinogenesis through formation of chimeric or fusion genes via translocations, amplifications, and rearrangements. Because these fusion genes are by nature specific to cancer, they may represent promising drug targets or diagnostic reagents, as exemplified by BCR-ABL fusion genes in chronic myelogenous leukemia or EML4-ALK gene fusions in lung cancer. Recently, our group has reported the pioneering discovery of fusion genes in gastric cancer. Working with a group in Michigan, we identified a rare gene fusion involving the RAF kinase in a small percentage of gastric cancers. Importantly, cancer cell lines expressing these fusions were sensitive to treatment by sorafenib, a RAF small molecule inhibitor. This year, we discovered another recurrent gene fusion in gastric cancer involving the coding region of the SLC1A2 gene to CD44 regulatory elements in gastric cancer cell lines and human gastric resection samples. Experimentally, this fusion gene resulted in a pro-oncogenic metabolic milieu favoring tumor growth and survival. This discovery provides further evidence that recurrent gene fusions, first described in hematological malignancies might also be important in solid organ cancers.
Microsatellite Instability (MSI) is characterized by widespread replication errors in simple repetitive microsatellite sequences because of defects in mismatch repair genes. First demonstrated in colon cancer and generally associated with improved prognosis, MSI has been shown to be associated with intestinal-type gastric tumors. One consequence of MSI is the production of oncogenic mutations in the EGFR-MAPK and PI3K pathways for example MLK3, a gene that encoding mixed lineage kinase 3, a kinase involved in MAPK signaling, is commonly mutated in MSI cancers. Another pathway known to be associated with MSI is the TGF-ß tumor suppressor pathway, where genetic alterations caused by MSI can inactivate the TGF-ß Type II receptor.
Besides alterations that affect DNA nucleotide sequence and copy number, epigenetic alterations such as DNA methylation and histone acetylation/methylation can also exert a profound impact on gastric cancer development. Aberrant methylation patterns with genome-wide hypomethylation occurring concomitantly with abnormal hypermethylation of specific genes occurs relatively commonly in gastric cancer. Functionally, the hypermethylation of CpG islands in gene promoters can cause the transcriptional silencing of tumor suppressor genes. Epigenetic differences between the two subtypes of gastric cancer have been reported. hMLH1 inactivation by methylation is associated with the intestinal subtype whilst CDKN2A inactivation by methylation is more frequently described in the context of diffuse type cancers. Additionally, a CpG island methylator phenotype (CIMP) referring to a subset of Gastric cancers harboring concordant methylation of multiple promoter CpG island loci has been described. CIMP-high Gastric cancers tend to be of the Diffuse subtype, have advanced stage and poor prognosis. CIMP is also characteristic of Ebstein Barr Virus-associated Gastric cancer which accounts for 10% of all gastric cancers.
Recent discoveries have implicated a new class of noncoding RNA known as microRNAs (miRNAs) in gastric cancer progression. miRNAs offer another dimension of precise genetic regulation (post-transcriptional regulation) by identifying specific sequences (called complementary sequences) on target messenger RNA transcripts (mRNAs), usually resulting in translational repression (prevention of generation of protein product) or target degradation and gene silencing. Our group and others have identified several miRNAs that demonstrate differential expression in gastric cancer tissues and also modulate pro-oncogenic traits in experimental models. In particular, these miRNAs have been shown to directly or indirectly regulate the expression or activity of proapoptotic and antiapoptotic members of Bcl-2 family. This has led to interest in developing miRNA-directed therapeutics although challenges of clinical delivery of these biologic molecules remain to be circumvented.
As described above, the Laurens subtypes are characterized by different epidemiology, morphology, pathogenesis, clinical behavior, and somatic aberrations. However, based on morphology, reproducibility of the Laurens classification among pathologists is only about 70%, limiting its clinical applicability. Recently, we attempted to move beyond Laurens classification and employed unsupervised genomic techniques to identify 2 major subtypes of Gastric cancer (intrinsic subtypes) with very distinct gene expression patterns. Because these intrinsic subtypes were correlated with Laurens subclasses, we termed these subtypes Genomic Intestinal and Genomic Diffuse. Whilst correlated, it is important to emphasize that in univariate and multivariate analyses in multiple cohorts, the intrinsic genomic subtypes were prognostic of survival, but Laurens histological subtypes were not. These genomic subtypes may thus refine identification of the 2 major classes of gastric cancer and improve upon Laurens by providing prognostic information for risk stratification. Furthermore, the 2 subtypes exhibit distinct response patterns to standard cytotoxics providing a potential to develop subtype-specific therapeutic strategies based on molecular vulnerabilities to drugs.
In conclusion, our knowledge about the molecular genetics of gastric cancer, both in terms of germline susceptibility and somatic molecular aberrations is accumulating rapidly. Further insights into molecular makeup of this important disease will emerge from the anticipated torrent of molecular information following rapid advances in -omic technologies and co-ordinated international endeavors including the Cancer Genome Atlas. When interpreting the data, it is useful to consider Gastric cancer, not as one disease, but at least 2 major subtypes with unique features.
 Click here to download the full issue for USD 6.50 Click here to download the full issue for USD 6.50
|
